Please try it: NGphylogeny.fr.
Documentation
Table of Content
- 1. Overview
- 2. Phylogeny Analysis
- 3. Blast: Sequence explorer
- 4. Programs use and technical details
- 5. Formats
- 6. More information about external softwares
1. Overview
Phylogeny.fr has been designed to provide a high performance platform that transparently chains programs relevant to phylogenetic analysis in a comprehensive, and flexible pipeline. Although phylogenetic aficionados will be able to find most of their favorite tools and run sophisticated analysis, the primary philosophy of Phylogeny.fr is to assist biologists with no experience in phylogeny in analyzing their data in a robust way.
The Phylogeny.fr platform offers a phylogeny pipeline which can be executed through three main modes:
The "One Click mode" targets users that do not wish to deal with program and parameter selection. By default, the pipeline is already set up to run and connect programs recognized for their accuracy and speed (MUSCLE for multiple alignment and PhyML for phylogeny) to reconstruct a robust phylogenetic tree from a set of sequences.
In the "Advanced mode", the Phylogeny.fr server proposes the succession of the same programs but users can choose the steps to perform (multiple sequence alignment, phylogenetic reconstruction, tree drawing) and the options of each program.
The "A la carte mode" offers the possibility of running and testing more alignment and phylogeny programs, such as MUSCLE, ClustalW, T-Coffee, PhyML, BioNJ, TNT,...
Alternatively, users have the possibility to run the different programs separately.
↑ Table of Content2. Phylogeny Analysis
2.1 "One Click" mode
This is a "default" mode which proposes a pipeline already set up to run and connect programs recognized for their accuracy and speed (MUSCLE for multiple alignment, optionally Gblocks for alignment curation, PhyML for phylogeny and finally TreeDyn for tree drawing) to reconstruct a robust phylogenetic tree from a set of sequences.
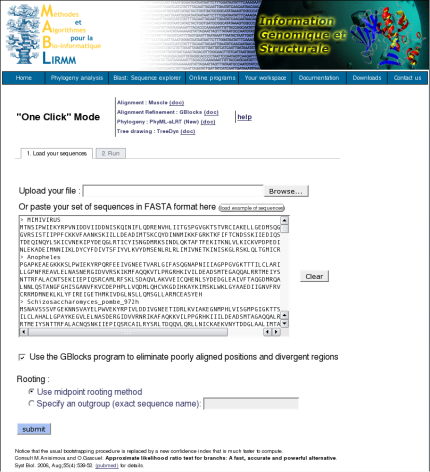
What users have to do is just to copy and paste their set of sequences in the FASTA format (or upload their FASTA file) and to click the Submit button. The system will do all the rest work, all the parameters are those of programs by default. However, users are able to decide to use or not the Gblocks program to eliminate poorly aligned positions and divergent regions, by checking the corresponding checkbox in the form page. At the end of the analysis, the server displays a publication quality image of the phylogenetic tree.
| Step | Program used | Settings | Notes |
|---|---|---|---|
| Alignment | MUSCLE 3.7 |
|
Several studies and especially the BAliBASE benchmark showed that MUSCLE achieved the highest ranking of any method at the time of publication. |
| Alignment refinement | Gblocks 0.91b |
|
This step is optional. Gblocks eliminates poorly aligned positions and divergent regions (removes alignment noise). Parameters are set to their default values in Gblocks. These are rather stringent; e.g. all positions with gaps are removed. |
| Phylogeny | PhyML 3.0 |
|
PhyML was shown to be at least as accurate as other existing phylogeny programs using simulated data, while being one order of magnitude faster. HKY fits well in most cases as it modelizes the main features of DNA substitutions: transition/transversion and unequal base frequencies. LG has been shown to be the best amino-acid replacement matrix to date. Gamma distributed rates are mandatory in most (if not all) analyses, and using invariant sites generally improves (never degrade) the fit. |
| Tree rendering | TreeDyn 198.3 |
|
TreeDyn offers many tree customization options compared to other tree rendering tools and especially for tree annotations. The starting output tree is rooted using mid-point rooting method (performed by Retree from PHYLIP package) but the user can reroot the tree using our dynamic tree editing interface. |
↑ Table of Content
2.2 Advanced mode
The pipeline is the same as the "One Click" mode but is flexible
enough to allow users to select which steps to perform. In this manner, the
input data can be a set of non-aligned sequences in FASTA format, an
alignment of multiple sequences in FASTA, PHYLIP or Clustal format, or a
tree in NEWICK format.
Users are provided with options to set the parameters of the different
programs of the pipeline.
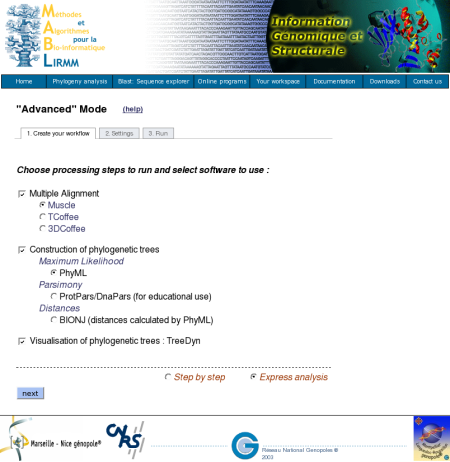
Furthermore, the system offers the possibility to control results of each step before launching the next program, so that users can modify and properly adjust parameters for a given task. This is possible in checking the "Step by step" option.
In the case of an "All at once", at the end of the pipeline processing, users have access to detailed reports for all the different analysis steps of the pipeline through different tabs of results.
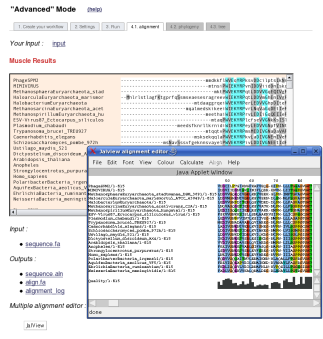
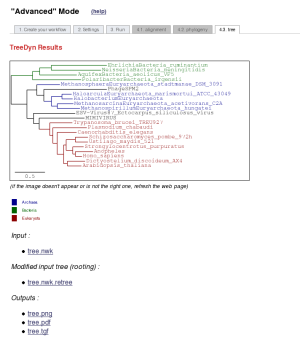
At the end of the analysis, users have the possibility to look at a schematic representation of the workflow, with details about software options, so as the references of the different selected tools. This summary of the run is accessible through the "Overview" tab since the analysis is finished. This fonctionality is also observable in the "One Click mode".
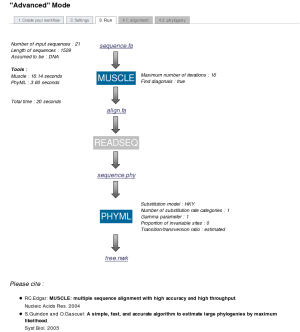
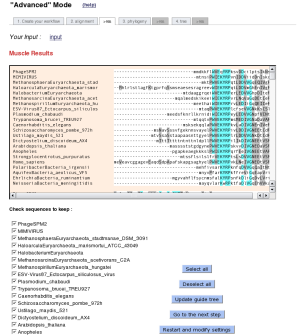
In the case of an "Step by step" analysis, users have the possibility to control and edit the results of each step before launching the next program.
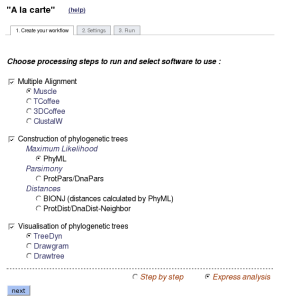
2.3 "A la carte" mode
The server offers the possibility to run other alignment and phylogeny programs than those preselected in the "One Click" and "Advanced" modes:
- a multiple sequence alignment using MUSCLE, T-Coffee, 3DCoffee or ClustalW according to user preference
- a phylogenetic reconstruction using PhyML, TNT, BioNJ or Neighbor and
- the generation of a publication quality image of the resulting phylogenetic tree using TreeDyn, Drawgram or Drawtree
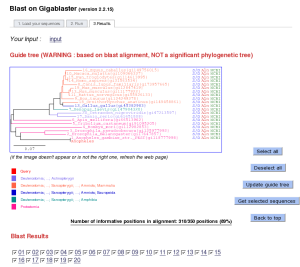
3. Blast: Explore your sequence neighbors
A fast BLAST search on Gigablaster allows to quickly explore your sequence neighbors. Paste your single sequence, run blast and explore its homologous sequences. The system facilitates the selection of homologous sequences, based on a 'quick-and-dirty' phylogenetic representation using BLAST results and an estimator of the final multiple alignment length.
4. Programs use and technical details
| Program | Function | Input | Output | Speed | Current Limitations | Use |
|---|---|---|---|---|---|---|
| Blastall 2.2.17 | Sequence searching | Raw, FASTA | FASTA | Fast | None | Advanced and "A la Carte" |
| MUSCLE 3.7 | Multiple alignment | FASTA, EMBL/Uniprot, GenBank, PAUP*/Nexus | FASTA, Clustal, PHYLIP | Fast | <200 nucleic sequences, <6000 sites <200 protein sequences, <2000 sites |
All modes Large dataset |
| T-Coffee 5.56 | Multiple alignment | FASTA, EMBL/Uniprot, GenBank, PAUP*/Nexus | FASTA, Clustal, PHYLIP | Very slow | <50 nucleic sequences, <2000 sites <50 protein sequences, <2000 sites |
"A la Carte" Small dataset |
| 3DCoffee 5.56 | Multiple alignment using structural information | FASTA, EMBL/Uniprot, GenBank, PAUP*/Nexus | FASTA, Clustal, PHYLIP | Very slow | <50 nucleic sequences, <2000 sites <50 protein sequences, <2000 sites |
"A la Carte" Small dataset |
| ClustalW 2.0.3 | Multiple alignment | FASTA, EMBL/Uniprot, GenBank, PAUP*/Nexus | FASTA, Clustal, PHYLIP | Fast | <200 nucleic sequences, <4000 sites <200 protein sequences, <2000 sites |
"A la Carte" Large dataset |
| Gblocks 0.91b | Alignment refinement | FASTA, Clustal, PHYLIP, EMBL, PAUP*/Nexus | FASTA, Clustal, PHYLIP | Fast | None | All modes Large dataset |
| PhyML 3.0 | Phylogeny using maximum likelihood | FASTA, Clustal, PHYLIP, EMBL, PAUP*/Nexus | Newick | Fast to Slow | (sequence size) × (number of taxa)2 <100000000 | All modes Medium to large dataset |
| TNT 1.1 | Phylogeny using parsimony | FASTA, Clustal, PHYLIP, EMBL, PAUP*/Nexus | Newick | Fast to Slow | <200 sequences, <6000 sites | "A la Carte" Medium to large dataset |
| FastDist/Protdist + BioNJ / Neighbor (PHYLYP) 3.67 |
Phylogeny using distances | FASTA, Clustal, PHYLIP, EMBL, PAUP*/Nexus | Newick | Fast | <5000 taxa for BioNJ <500 taxa for Neighbor |
"A la Carte" Large dataset |
| Bootstrap with PhyML and TNT | Estimations of clade supports | FASTA, Clustal, PHYLIP, EMBL, PAUP*/Nexus | Newick | Very slow | <500 replicates with PhyML <1000 replicates with TNT |
Advanced and "A la Carte" Small to medium datasets |
| Bootstrap with distance methods | Estimations of clade supports | FASTA, Clustal, PHYLIP, EMBL, PAUP*/Nexus | Newick | Slow | <1000 replicates with FastDist <1000 replicates with Protdist |
"A la Carte" Medium to large datasets |
| TreeDyn 198.3 | Tree rendering | Newick, New Hampshire Extanded, PAUP*/Nexus | Newick, PNG, PS, PDF, SVG, TGF | Fast | None | All modes |
| Drawgram 3.67 | Various tree shapes rendering | Newick, New Hampshire Extanded, PAUP*/Nexus | PNG, PS, PDF | Fast | None | "A la Carte" |
| Drawtree 3.67 | Unrooted tree rendering | Newick, New Hampshire Extanded, PAUP*/Nexus | PNG, PS, PDF | Fast | None | "A la Carte" |
↑ Table of Content
5. Formats
5.1 FASTA format
This format can be used for single sequence, multiple sequences or alignment. Each sequence begins with a single header line providing the sequence name (optionnaly description), followed by lines of sequence data. The description line must start with a greater-than (">") symbol in the first column.
>Homo sapiens MEVEAVCGGAGEVEAQDSDPAPAFSKAPGSAGHYELPWVEKYRPVKLNEIVGNEDTVSRLEVFAREGNVP NIIIAGPPGTGKTTSILCLARALLGPALKDAMLELNASNDRGIDVVRNKIKMFAQQKVTLPKGRHKIIIL DEADSMTDGAQQALRRTMEIYSKTTRFALACNASDKIIEPIQSRCAVLRYTKLTDAQILTRLMNVIEKER VPYTDDGLEAIIFTAQGDMRQALNNLQSTFSGFGFINSENVFKVCDEPHPLLVKEMIQHCVNANIDEAYK ILAHLWHLGYSPEDIIGNIFRVCKTFQMAEYLKLEFIKEIGYTHMKIAEGVNSLLQMAGLLARLCQKTMA PVAS >Arabidopsis thaliana MASSSSTSTGDGYNEPWVEKYRPSKVVDIVGNEDAVSRLQVIARDGNMPNLILSGPPGTGKTTSILALAH ELLGTNYKEAVLELNASDDRGIDVVRNKIKMFAQKKVTLPPGRHKVVILDEADSMTSGAQQALRRTIEIY SNSTRFALACNTSAKIIEPIQSRCALVRFSRLSDQQILGRLLVVVAAEKVPYVPEGLEAIIFTADGDMRQ ALNNLQATFSGFSFVNQENVFKVCDQPHPLHVKNIVRNVLESKFDIACDGLKQLYDLGYSPTDIITTLFR IIKNYDMAEYLKLEFMKETGFAHMRICDGVGSYLQLCGLLAKLSIVRETAKAP
5.2 GenBank format
This format can be used for single sequence or multiple sequences.
Each sequence entry begins with a line containing the word "LOCUS",
indicating the short name for this sequence, followed by several annotation
lines. The start of each sequence is marked by a line containing the word
"ORIGIN" and the end of each sequence is marked by two slashes
("//").
LOCUS CAA36839 152 aa linear PRI 14-NOV-2006
DEFINITION calmodulin [Homo sapiens].
ACCESSION CAA36839
VERSION CAA36839.1 GI:825635
DBSOURCE embl accession X52606.1
embl accession X52607.1
embl accession X52608.1
ORIGIN
1 madqlteeqi aefkeafslf dkdgdgtitt kelgtvmrsl gqnpteaelq dminevdadd
61 lpgngtidfp efltmmarkm kdtdseeeir eafrvfdkdg ngyisaaelr hvmtnlgekl
121 tdeevdemir eadidgdgqv nyeefvqmmt ak
//
LOCUS CAA59418 149 aa linear PLN 18-APR-2005
DEFINITION calmodulin [Macrocystis pyrifera].
ACCESSION CAA59418
VERSION CAA59418.1 GI:728609
DBSOURCE embl accession X85091.1
ORIGIN
1 madqlteeqi aefkeafslf dkdgdgtitt kelgtvmrsl gqnpteaelq dminevdadg
61 ngtidfpefl tmmarkmkdt dseeeiieaf kvfdkdgngf isaaelrhim tnlgekltde
121 evdemiread idgdgqinye efvkmmmak
//
5.3 EMBL format
This format can be used for single sequence or multiple sequences.
Each sequence starts with an identifier line containing the word
"ID ", followed by several annotation lines. The start of each
sequence is marked by a line starting with the identification "SQ",
and the end of each sequence is marked by two slashes ("//").
ID Homo sapiens; AA; UNK; 354 AA.
XX
AC unknown;
XX
DE
XX
FH Key Location/Qualifiers
FH
XX
SQ Sequence 354 BP; 35 A; 8 C; 23 G; 18 T; 270 other;
meveavcgga geveaqdsdp apafskapgs aghyelpwve kyrpvklnei vgnedtvsrl 60
evfaregnvp niiiagppgt gkttsilcla rallgpalkd amlelnasnd rgidvvrnki 120
kmfaqqkvtl pkgrhkiiil deadsmtdga qqalrrtmei yskttrfala cnasdkiiep 180
iqsrcavlry tkltdaqilt rlmnvieker vpytddglea iiftaqgdmr qalnnlqstf 240
sgfgfinsen vfkvcdephp llvkemiqhc vnanideayk ilahlwhlgy spediignif 300
rvcktfqmae ylklefikei gythmkiaeg vnsllqmagl larlcqktma pvas 354
//
ID Arabidopsis thaliana; AA; UNK; 333 AA.
XX
AC unknown;
XX
DE
XX
FH Key Location/Qualifiers
FH
XX
SQ Sequence 333 BP; 27 A; 6 C; 21 G; 18 T; 261 other;
masssststg dgynepwvek yrpskvvdiv gnedavsrlq viardgnmpn lilsgppgtg 60
kttsilalah ellgtnykea vlelnasddr gidvvrnkik mfaqkkvtlp pgrhkvvild 120
eadsmtsgaq qalrrtieiy snstrfalac ntsakiiepi qsrcalvrfs rlsdqqilgr 180
llvvvaaekv pyvpegleai iftadgdmrq alnnlqatfs gfsfvnqenv fkvcdqphpl 240
hvknivrnvl eskfdiacdg lkqlydlgys ptdiittlfr iiknydmaey lklefmketg 300
fahmricdgv gsylqlcgll aklsivreta kap 333
//
5.4 PHYLIP format
This is an alignment format.
The first line contains the number of sequences and their length (in
characters) separated by blanks.
The next line contains the sequence name, followed by the sequence in blocks
of 10 characters.
7 100
T25 ACTATTGAAA GAAGGGGGTT CCTAGATATC TGCGAGTATA ATCGTGCTTG
T16 ATTAATCAAA GTAGGCGGGG CGGCCGTAGA TGCTAAGAAA ATCGAGTTCG
T27 ATTAATCAAA GTAGGCAGGG CGGCCGTAGA TGCTAAGAAA ATCGAGTTCG
T1 GTTAACCGAA GTAGGCGGAA CGGACGTATA TGCGATTAAA ATCGAGTTCG
T19 GTTAACCGAA GTAGGCGGAA CGGACGTATA TGCGATTAAA ATCGAGTTCG
T35 ATTAATCAAA GCAGGCGGTC CGGACGTATA TCCTAATAAA ATCGAGTTCG
T56 ATTAATCAAA GTAGGCGGTC CGGCCGAATA TGCGAATAAA ATCGAGTTCG
GTCTCCTATC GATGCGCATC GGACCGAGAG GCTCTCCAGC CATGTGGACG
GTCACCTCCC ATTGGGCAGC AGATCGCTAG GCTCTTTAGC CAGGTGGACG
GTCACCTCCC ATTGCGCAGC AGATCGCTAG GCTCTTTAGC CAGGCGGACG
GACACCTTCC AGGGCGCAGC AGATCGCGAG GCTTTCTAAC CAGGTGGACG
GACACCTTCC AGGGCGCAGC AGATCGCGAG GCTTTCTAGC CAGGTGGACG
GTCACCTCCC AGGGCGCAGA AGATCGCGAG GCTCTCCAGC CAGGTGGACG
GTAACCTCCC AGTCCGCAGA AGATCGCGAG GCTCTCCAGC CAGGGGGACG
5.5 Clustal format
This is an alignment format.
The word "CLUSTAL" is on the first line of the file. The alignment
is written in blocks of a fixed length. Every block starts with the sequence
names (maximum of 10 characters), followed by at least one space character.
The sequence is then displayed in upper or lower cases, "-" denotes
gaps.
A count of the total number of residues may be shown at the end of the line.
Below each block of residues, an additional line shows the degree of conservation for each site.
CLUSTAL W (1.83) multiple sequence alignment
aboA -NLFV-ALYDFVASGDNTLSITKGEKLRV-------LGYNHNG-------EWCEA--QTK 42
ycsB KGVIY-ALWDYEPQNDDELPMKEGDCMTI-------IHREDEDEI-----EWWWA--RLN 45
pht -GYQYRALYDYKKEREEDIDLHLGDILTVNKGSLVALGFSDGQEARPEEIGWLNGYNETT 59
vie ---------DRVRKKSG--AAWQGQIVGW---------YCTNLTP----EGYAVESEAHP 36
ihvA ------NFRVYYRDSRD--PVWKGPAKLL---------WKGEG-------AVVIQ---DN 33
. *
aboA NGQGWVPSNYITPVN------ 57
ycsB DKEGYVPRNLLGLYP------ 60
pht GERGDFPGTYVEYIGRKKISP 80
vie GSVQIYPVAALERIN------ 51
ihvA SDIKVVPRRKAKIIRD----- 49
. *
5.6 Nexus
Nexus format can be used for multiple sequences, alignments, distance matrices and trees. It starts with "#NEXUS". It has been detailed in Maddison & al., NEXUS: An Extensible File Format for Systematic Information.
#NEXUS BEGIN TAXA; DIMENSIONS ntax=5; TAXLABELS 1aboA 2ycsB 3pht 4vie 5ihvA; END; BEGIN UNALIGNED; DIMENSIONS ntax=5; FORMAT datatype=Protein gap=-; MATRIX 1aboA NLFVALYDFVASGDNTLSITKGEKLRVLGYNHNGEWCEAQTKNGQGWVPSNYITPVN 2ycsB KGVIYALWDYEPQNDDELPMKEGDCMTIIHREDEDEIEWWWARLNDKEGYVPRNLLGLYP 3pht GYQYRALYDYKKEREEDIDLHLGDILTVNKGSLVALGFSDGQEARPEEIGWLNGYNETTGERGDFPGTYVEYIGRKKISP 4vie DRVRKKSGAAWQGQIVGWYCTNLTPEGYAVESEAHPGSVQIYPVAALERIN 5ihvA NFRVYYRDSRDPVWKGPAKLLWKGEGAVVIQDNSDIKVVPRRKAKIIRD ; END;
6. More information about external softwares
- Multiple Alignment:
- Phylogeny:
- Tree Viewers:
- Alignment Refinement:
↑ Table of Content